
Patient & Advocacy Resources

Supporting Patients & Families
We welcome new partnerships with:
Aerys Biosciences is dedicated to improving the lives of people affected by idiopathic pulmonary fibrosis (IPF) and pulmonary arterial hypertension (PAH). These are progressive, life-altering conditions that impose a heavy physical, emotional, and financial burden which affects not only on patients, but also their families and caregivers.
For IPF and PAH, symptoms are often subtle at first, diagnoses can be delayed, and treatment options remain limited. At Aerys Biosciences, we have spoken with patients and understand that they deserve clear information, better therapies, and partners who are committed to advancing meaningful solutions.
That is why we aim to be more than a biopharmaceutical company and we work to be a trusted partner to bring effective therapies to the clinic. Through innovative research, patient-centered clinical programs, and strategic collaborations with advocacy groups, clinicians, and research networks, we strive to deliver new hope through next-generation small-molecule therapies.
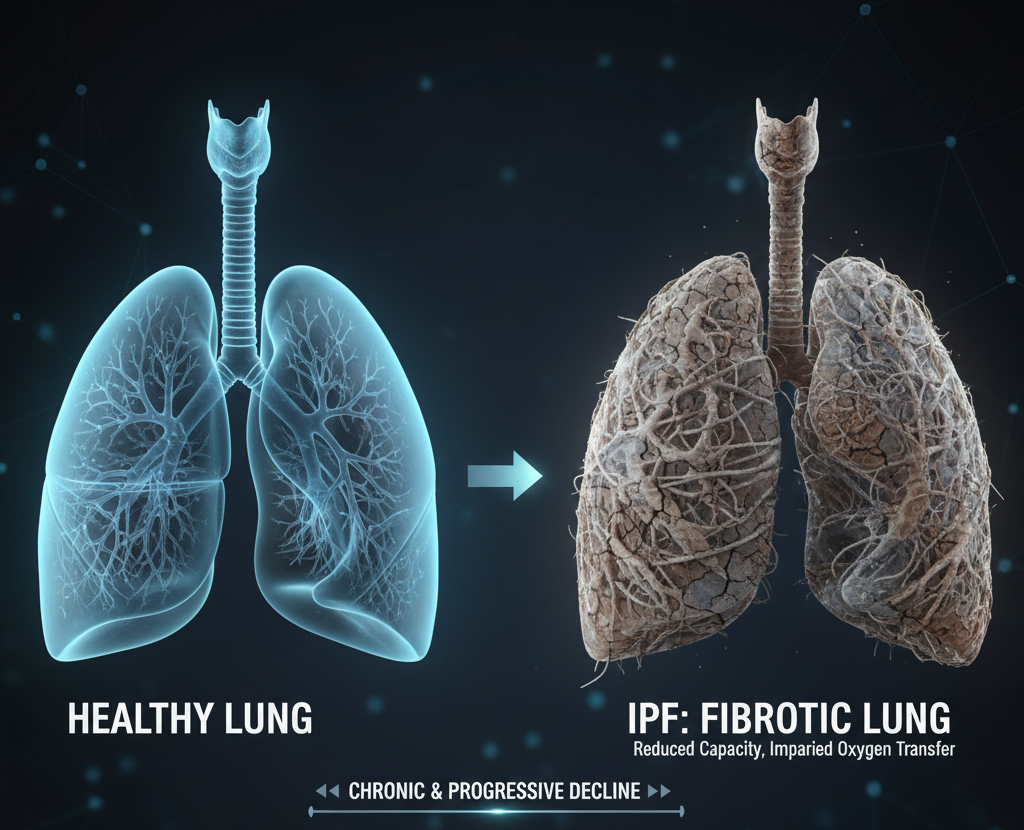
Idiopathic Pulmonary Fibrosis(IPF)
Idiopathic Pulmonary Fibrosis (IPF) is a chronic, progressive lung disease characterized by scarring of the lung tissue. Over time, this fibrosis reduces lung capacity and impairs oxygen transfer, leading to worsening shortness of breath, fatigue, and functional decline.
Unlike inflammation-driven lung conditions that may improve with treatment, IPF continues to worsen even with currently approved therapies. As scarring accumulates, patients face increasing respiratory limitations and frequent hospitalizations.
- Persistent shortness of breath
- Dry, hacking cough
- Fatigue and reduced exercise tolerance
- Unintentional weight loss
- Digital clubbing (rounding of fingertips)
The cause of IPF remains unknown, but contributing factors may include:
- Genetic predisposition
- Environmental exposures (dust, smoke, metal particulates)
- Viral infections
- Age-related cellular dysfunction
- Aberrant fibroblast activation and extracellular matrix remodeling
- Anti-fibrotic medications that slow—but do not stop—progression
- Oxygen therapy
- Pulmonary rehabilitation
- Lung transplant evaluation for eligible patients
- Supportive care to manage symptoms and improve daily functioning
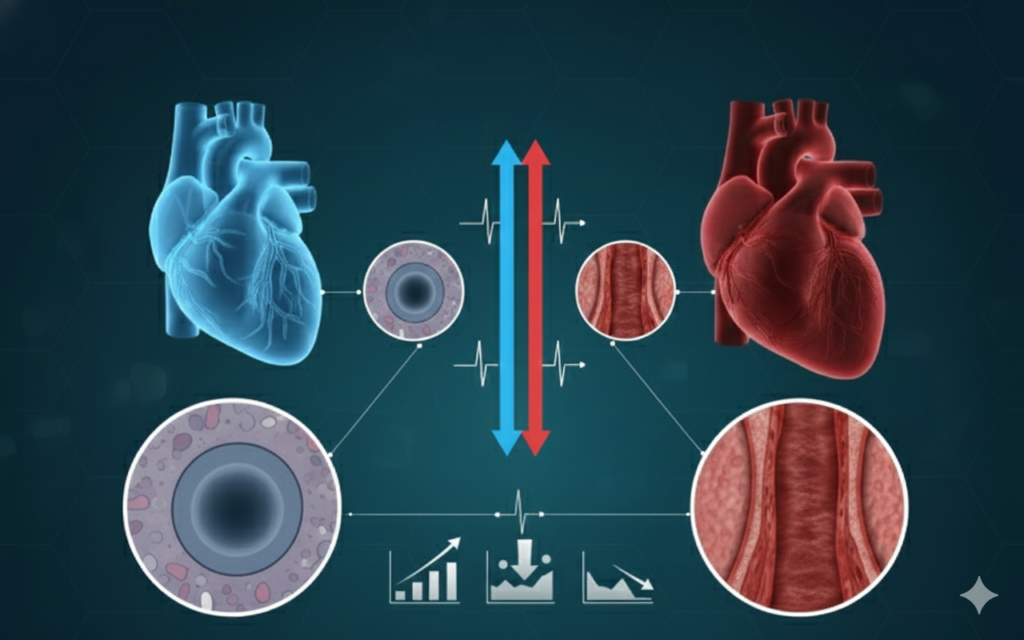
Pulmonary Arterial Hypertension (PAH)
Pulmonary Arterial Hypertension (PAH) is a severe, progressive condition marked by narrowing and stiffening of the pulmonary arteries. This remodeling increases pressure in the lung circulation, overloading the right side of the heart and eventually leading to right-heart failure if untreated.
PAH can occur on its own or in association with connective tissue diseases, congenital heart disease, HIV, or certain drugs/toxins. Despite advances in therapy, many patients experience worsening symptoms, limited exercise capacity, and reduced quality of life.
- Shortness of breath—especially during activity
- Chest pain or pressure
- Lightheadedness or fainting
- Fatigue and weakness
- Swelling in the ankles, legs, or abdomen
PAH may result from:
- Genetic variants affecting vascular tone (e.g., BMPR2)
- Endothelial dysfunction and inflammation
- Abnormal smooth muscle proliferation
- Chronic fibro-inflammatory signaling in the pulmonary arteries
- Vasodilators targeting nitric oxide, endothelin, or prostacyclin pathways
- Combination therapy for disease control
- Diuretics and supportive medications
- Exercise and lifestyle adjustments
- Lung or heart–lung transplantation in advanced cases